Currently, world-wide ~44 million people have Alzheimer’s disease (AD). With an ageing population, this number is expected to rise and costs for care could reach $1.1 trillion by 2050. Since recent clinical trials have failed to prevent the progression of the disease, a better understanding of the toxic molecular features that cause neuronal cell death is urgently required. A recent paper published in Nature by Lee et. al1 have now identified an increase in somaticDNA recombination in certain Alzheimer’s patients of a protein associated with Alzheimer’s,APP. Not only does this landmark paper reveal a whole new side to AD biology but is also provides the first evidence that DNA recombination can occur in thebrain.
What is Alzheimer’s disease?
Most peopleknow that Alzheimer’s disease (AD) is a neurodegenerative disease and the most common cause of dementia, however the molecular characteristics and even moreso, the cause of AD, is little understood. The disease is characterised by thedeposition of extracellular plaques of the protein beta-amyloid (Aβ) and intracellular tangles of a protein, tau. Symptoms associated with these characteristics include short-term memory loss, progressing over time to include disorientation, behavioural changes and communication issues.

The Aβ plaques derive from a transmembrane precursor protein, literally, the amyloid precursor protein (APP) which when cleaved at both its N and C-terminus, by β- and γ-secretase, respectively, generates a 1-40, 1-42 or 1-43 fragment (Figure 1). The length of the fragment, determined by γ-secretase cleavage, has important consequences. The 1-42 long fragment is present in higher levels in AD patients and can result in plaque formation.
Cases of AD can be split into two groups; those who inherit genetic predispositions todevelop AD (familiar AD, or FAD) and those who develop it sporadically –sporadic AD, or SAD. The kind of genetic predispositions seen in patients withFAD include mutations and duplications in the gene encoding APP. Since the APPis found on chromosome 21, three copies of the gene are found in people with Down Syndrome. These copy number variations (CNVs) of APP have a causal link to AD. However, the majority of AD cases are SAD. The greatest risk of SAD is age;whilst this offers a sad future, it also suggests that it may be possible toalleviate the development of AD earlier on to prevent full progression of the disease. However, to achieve this, a complete understanding of molecular basis ofAD is needed.
The molecular basis of the APP gene
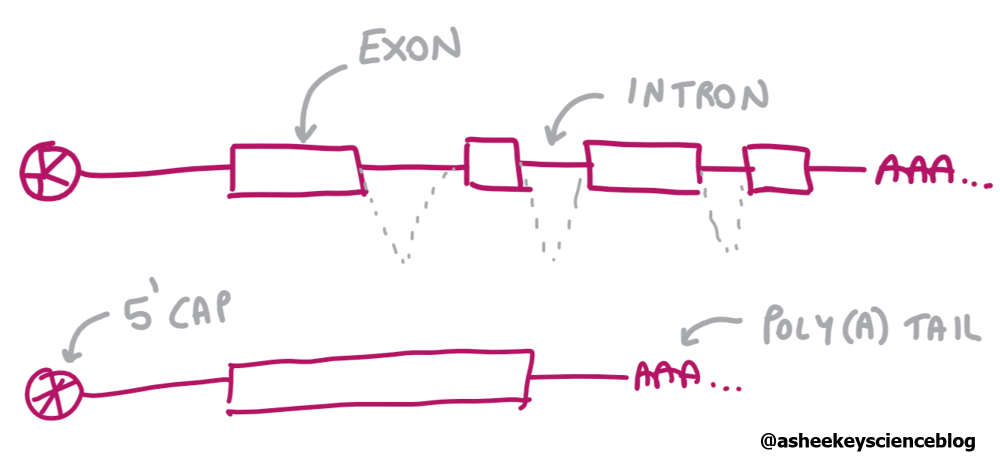
When genes are transcribed they contain coding (exons) and non-coding regions (introns) (Figure 2,3). After processing, a combination of exons remain. This mRNA with only exons can be “reverse-transcribed” to DNA – this DNA fragment of only the coding parts of a gene is referred to as cDNA. The APP contains 18 exons, however, alternative copies of the gene can be made with different combination of the exons (splice variants).
Lee’s team had previously identified CNVs of APP in neurons that showed differences in the combination of exons present. To explore this further, the authors used a protocol that enabled them to take nuclei from neurons, extract RNA, reverse transcribe the RNA to DNA and amplify (RT-PCR) only APP mRNA sequences. After running the products on a gel to separate them by size, the fragments were then probed to identify the APP transcripts. Identified fragments were then cloned, sequenced and mapped onto the complete APP gene.
In addition to expected splice variants, the authors noticed that there were multiple unexpected fragments seen in the gels from SAD patients compared to non-diseased control samples. Sequencing showed these unexpected fragments to show novel exon junctions, known as intra-exonic junctions (IEJs) – many of these fragments were missing central exons and hence if translated, would give rise to truncated/altered versions of APP. A common IEJ was between the 24th nucleotide of exon 3 and the 45th nucleotide of exon 16 (R3/16). Many of these fragments were seen when APP gene fragments were amplified from DNA samples, supporting the notion that the mosaic-exon APP mRNA seen are encoded as genomic DNA (gencDNAs). Moreover, in a mouse model, the presence of one gencDNA showed an age-dependent increase. Unexpected fragments were not seen though when the team studied another AD-related gene, PSEN1.
Why are more APP gencDNAs found in SAD patients?
Using a fluorescent-imaging technique, the team saw that the gencDNAs did not co-localise with the wild-type locus of APP. So, how did the gencDNAs arise? Using a Chinese hamster ovary cell line (as it has high levels of reverse transcriptase activity) expressing the full-length APP gene (without introns), Lee’s team showed that gencDNA formation was enhanced when DNA strand breaks were induced and greatly reduced when a reverse transcriptase inhibitor was present. This supported a mechanism by which gencDNAs derive from RNA transcripts and insert to damaged DNA sites. What predisposes SAD patients to this mechanism is not yet clear.
What’s the link with Alzheimer’s?
Since cleaved APP protein is attributed to plaque formation, it is possible that the gencDNAs, which represent only parts of the full APP gene, could be a source of pre-cleaved APP and thus enhance Aβ plaque formation. Alternatively, the age-dependent accumulation of gencDNA could create genomic instability and neuronal cell death. However, considering APP gencDNAs can be found in non-diseased patients, albeit much less of them, it suggests there may be a physiological role for their formation. As gencDNAs are in a different genomic location to the wild-type gene, expression regulation could be completely different and thus could aid synaptic function.
Does gene recombination occur in other cell types? How is gene recombination regulated? Are there other genes that form gencDNAs? This exciting study has unleashed a wealth of questions whose answers will be relevant to other brain physiologies besides AD. Nevertheless, the results give new hope to AD treatment. For example, if gencDNAs are found to be causal for SAD, regulation of gene recombination could become a new step for drug intervention in the hope to prevent the development of SAD. It would also be interesting to see how APP gencDNAs vary under different genetic backgrounds, since this may elucidate the reverse transcriptase and other proteins involved in this somatic genetic recombination mechanism.
Further reading
1. Lee, M. et al. SomaticAPP gene recombination in Alzheimer ’ s disease and normal neurons. Nature(2018). doi:10.1038/s41586-018-0718-6
